

The timsTOF SCP for quantitative single cell biology research with unbiased, deep single-cell 4D-Proteomics™, immunopeptidomics, epiproteomics and PTM analysis to complement scRNA-seq. Expanding the horizons of single cell research.
Expanding the horizons of single cell research
Redefining single cell proteomics
Advanced ion optics and PASEF® to investigate cell heterogeneity and biology from a single cell
Mass spectrometric proteomics has become a staple of modern research in understanding biological function and disease mechanisms. Healthy or diseased tissues that seem homogenous are composed of cells with a variety of different proteomes. The challenge of deciphering the proteomes in each single cell – the cell heterogeneity – holds the key to fully understanding its function.
The timsTOF SCP offers a radically improved ion source concept. Combined with parallel accumulation serial fragmentation (PASEF®) acquisition methods, it provides extremely high speed and sensitivity to tackle proteomes of single cells or post translational modifications in a few cells that are morphologically or functionally similar.
Radically improved ion transfer

The timsTOF SCP features a modified ion source geometry that includes 1 mm capillary identification for five times higher ion transfer into an additional higher pressure stage ion funnel and 8-stage differentially pumped vacuum system.
The larger capillary yields ultra-high sensitivity and the additional orthogonal ion reflection and subsequent funnel offers a separate, differentially pumped stage, maintaining the system robustness expected from the timsTOF instrument series.
Dramatic improvements in proteomics performance
The timsTOF SCP offers improvements in ion transmission into the source while maintaining robustness with an added higher pressure vacuum stage. This results in an almost 5x improvement in ion current. When combined with the Evosep One Whisper method running at 100 nL/min flow rate, and the dia-
PASEF® method, sensitivity gains of about 100X over previous high-flow results with the Evosep One are achieved. This enables unbiased proteomics at the true single cell level with good reproducibility, robustness and coverage of about 1500 proteins per cell for the first time.
Dual-TIMS, CCS-enabled analysis
Trapped ion mobility spectrometry (TIMS) is first and foremost a separation technique in the gas phase. This resolves sample complexity through an added dimension of separation in addition to high performance liquid chromatography (HPLC) and mass spectrometry, increasing peak capacity and confidence in compound characterization.
Equally important, the TIMS device also accumulates and concentrates ions of a given mass and mobility, enabling a unique increase in sensitivity and speed.
A near 100% duty cycle can be achieved with the dual-TIMS technology facilitating accumulation in the front section, while ions in the rear section are sequentially released depending on their mobility. This process of parallel accumulation serial fragmentation (PASEF®) enables collisional cross section (CCS) analysis.
CCS-enabled analysis opens up many further analytical possibilities, from greater certainty of compound identification to confident library matching and lower false discovery rates (FDRs) in large datasets.

Ideal for immunopeptidomics and other enrichment workflows
Besides unbiased true single cell proteomics applications, the timsTOF SCP also offers outstanding sensitivity for workflows that involve enrichment of peptides from the proteome. Immunopeptidomics studies start with purification of immunopeptides from plasma or tissue.
Since immunopeptides are present at relatively low abundance in these samples, the timsTOF SCP is ideal for immunpeptidomics for neo-antigen discovery where the available material is limited, as in needle biopsies. The timsTOF SCP also has the sensitivity to revolutionize the use of phosphoproteomics for the study of signaling pathways in cancer.
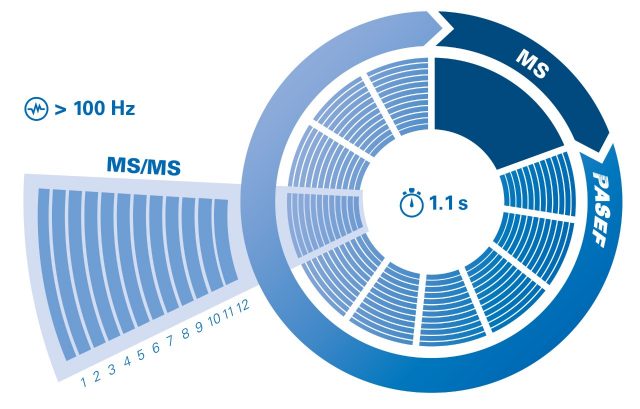
PASEF®
Peptide ions are separated using trapped ion mobility spectrometry (TIMS), eluted (~ 100 ms) and detected in the quadropole time of flight (QTOF), generating the TIMS MS heat map. In the PASEF® method the same TIMS separation is used with the quadrupole isolating a certain ion species during its elution and immediately shifting to the next precursor. Parent and fragment spectra are aligned by mobility values.
The parallel accumulation serial fragmentation (PASEF®) technology achieves a sequencing speed of >100 Hz. Using PASEF® increases the MS/MS spectra quality of the low abundant peptides by selecting them several times.
PASEF®: the perfect fit for shotgun proteomics
The timsTOF SCP powered by PASEF® offers a sequencing speed of >100 Hz without losing sensitivity or resolution. This is achieved by synchronizing the quadrupole isolation mass window with the elution time of the specific peptide packages from the TIMS funnel as well as the collision energy in the collision cell.
Bruker ProteoScape™ Run & Done – real time quality control for unbiased single cell analysis
The timsTOF SCP allows the analysis of hundreds of samples with minimal sample load (<200 ng) at sequencing speeds exceeding 100 Hz and with uncompromised proteomic depth. This has changed the way proteomics is studied, but the increased data requires a new level of data analysis.
Data analysis is a common bottleneck in many workflows. Modern analytical methods result in hundreds of lines of data generated by the timsTOF SCP. Bruker has introduced real-time database search capabilities - parallel database search engine in real-time (Bruker ProteoScape™) which removes this hurdle. Using Bruker ProteoScape™, as soon as a LC-MS run is completed, results are available – effectively Run & Done.
Advanced proteomics software processing
An open-file data format allows researchers to work directly with raw data and use industry leading software of their choice.
MaxQuant software has been adapted to manage 4-dimensional (4D) features in the space spanned by retention time, ion mobility, mass, and signal intensity. This benefits the identification and quantification of peptides, proteins, and posttranslational modifications, both from PASEF® and dia-PASEF® data.
PEAKS Studio combines de novo sequencing with traditional database searches and is optimized for processing timsTOF raw data.
The number of advanced proteomics processing software solutions reading timsTOF raw data is steadily growing, including Biognosys‘ Spectronaut and SpectroMine software as well as software from Skyline, Genedata, Protein Metrics, Mascot Distiller from Matrix Science, DIA-NN, and MSFragger.
Ultra-high sensitivity 4D-Proteomics™ with dia-PASEF®
CCS-enabled analysis for confident identifications
timsControl allows customization of the dia-PASEF® window scheme to focus on
the ions of interest. Adjusting the mass isolation width, TIMS range and cycle time allows adaptation of dia-PASEF® to different chromatography methods.
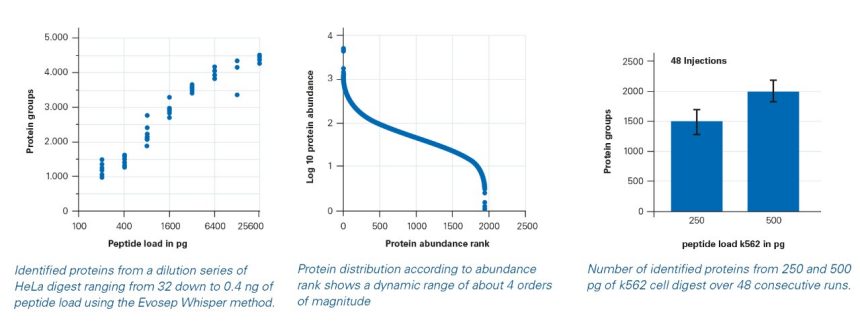
Combining low flow liquid chromatography on the Evosep One system with Whisper with the high sensitivity of dia-PASEF® on the timsTOF SCP, over 2000 proteins were identified from 500 pg of cell digest and 1500 proteins were identified from 250 pg, demonstrating the sensitivity needed for true single cell proteomics. The proteins identified from 250 pg covered an abundance range of about 4 orders of magnitude, enabling quantitative proteome analysis at single cell level.
Exploring the Tumor Microenvironment
Understanding the TME together with its infiltrating immune cells can be considered a crucial step to influence disease progression, treatment response and patient survival. Due to its high sensitivity the timsTOF SCP enables sufficient proteome depth on cells excised by laser capture microdissection (LMD) from FFPE tissue samples. A typical workflow is shown in the figure on the right.
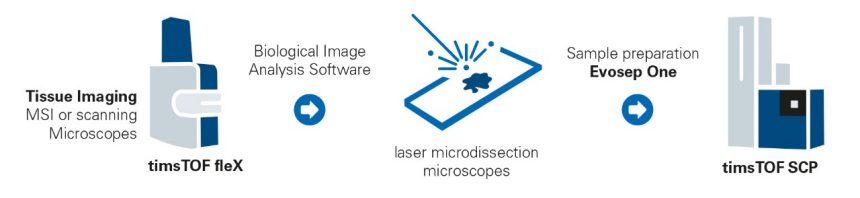
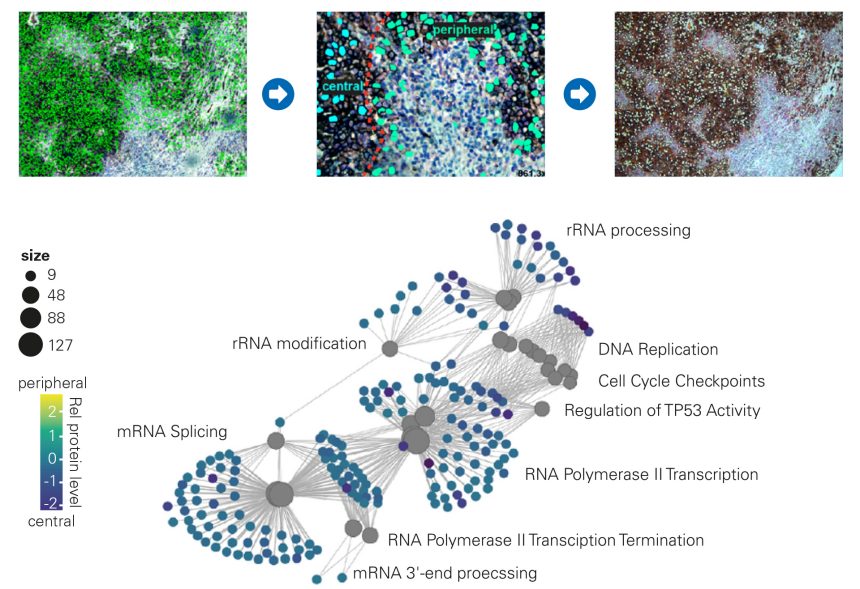
Cellular markers were used to identify melanoma cancer cells in the tumor mass and those closely related to the stroma for subsequent isolation of both populations by LMD and unbiased 4D-Proteomics™ analysis on a timsTOF SCP instrument.
Key Finding: Enrichment analysis revealed differentially regulated proteins between central and peripheral melanoma cells with potential for disease subtyping to guide clinical decision-making.
Results are provided by courtesy of Prof. Matthias Mann (doi: https://doi.org/10.1101/2020.12.22.423933)











